Certificiranje organizacij, Izobraževanje, Preskušanje in certificiranje proizvodov
03. avgust 2022


 Nazaj na vse novice
Nazaj na vse novice
Postopek certificiranja medicinskega pripomočka po Uredbi o medicinskih pripomočkih (EU) 2017/745
Pot za vstop medicinskega pripomočka na trg pelje od ideje, njegove proizvodnje in preskušanja do končne certifikacije. Priporočljivo je, da proizvajalec že v fazi
razvoja stopi v kontakt s priglašenim organom ter pridobi informacije o postopku certificiranja in zahtevah, ki jih medicinski pripomoček mora izpolnjevati.
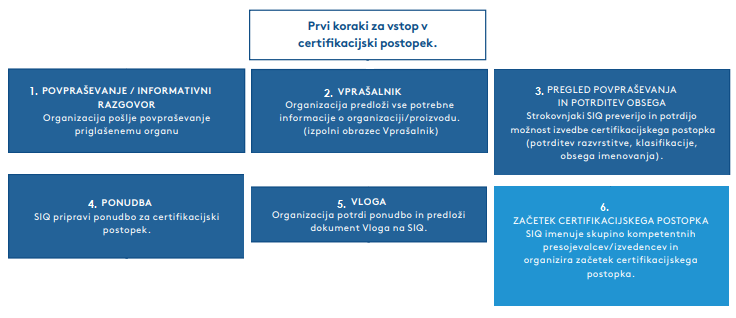
Slika 1.1.
Z uredbo MDR so zahteve za dokazovanje skladnosti medicinskega pripomočka strožje in obsežnejše. Celoten postopek preskušanja in certificiranja je dolgotrajen in lahko traja do dveh let, česar mnogi proizvajalci ne pričakujejo. Prvi koraki za vstop v certifikacijski postopek, ki segajo od prvega stika s stranko do potrditve ponudbe in načrtovanja presoje, so razvidni s slike 1.1.
In kako poteka certifikacijski postopek?
Organizacija priglašenemu organu predloži tehnično dokumentacijo. Ta izvede t. i. hiter pregled ali predpregled dokumentacije, kar pomeni, da preveri ustreznost strukture dokumentacije in prisotnost vseh zahtevanih dokumentov. Po potrditvi ustreznosti in popolnosti dokumentacije se certifikacijski postopek končno lahko prične.
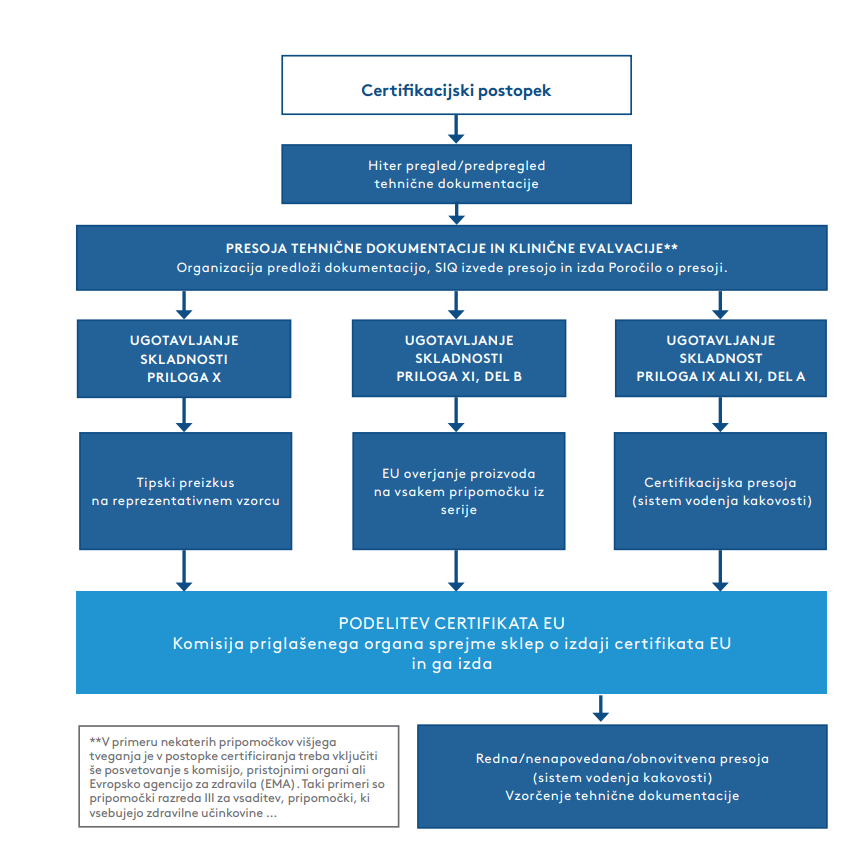
Prvi del postopka predstavlja presoja tehnične dokumentacije in klinične evalvacije proizvoda. SIQ v ta del vključuje vrsto usposobljenih strokovnjakov, tudi zdravnike specialiste in strokovnjake s posebnimi znanji (programska oprema, biokompatibilnost, sterilizacija …), kjer je to potrebno. Ko je presoja tehnične dokumentacije in klinične evalvacije zaključena in je dokumentacija v skladu z uredbo MDR, sledi nadaljevanje certifikacijskega postopka, in sicer glede na izbran postopek ugotavljanja skladnosti (Glej sliko 1.2.).
Najpogosteje se proizvajalci odločajo za postopek ugotavljanja skladnosti po Prilogi IX ali Prilogi XI, del A, ki vključuje presojanje vzpostavljenega sistema vodenja kakovosti. V takih primerih se certifikacijski postopek nadaljuje s certifikacijsko presojo. Presoja se lahko kombinira s presojo po standardu ISO 13485:2016. Ko organizacija odpravi vse neskladnosti in so vse zahteve izpolnjene, priglašeni organ izda certifikat (EU), s katerim lahko proizvajalci medicinskih pripomočkov vstopijo na trg EU.
Slika 1.2.
Treba je opozoriti, da v celotnem certifikacijskem postopku običajno največ časa vzame pregled tehnične dokumentacije in klinične evalvacije (prvi del presoje), kar lahko traja tudi od enega do dveh let.
Priglašeni organ v primeru ugotavljanja skladnosti po Prilogi IX ali Prilogi XI, del A, enkrat letno izvaja redne presoje certificiranega sistema vodenja kakovosti, vsako peto leto pa sledi obnovitvena presoja. Presojevalci na rednih in obnovitvenih presojah pregledujejo tudi poročila proizvajalca o spremljanju poprodajnih informacij o pripomočkih ter možne zaplete, ki jih imenujemo vigilančni primeri. Priglašeni organ najmanj enkrat v petih letih (razen v primeru medicinskega pripomočka razreda III, ko se presoja izvaja najmanj enkrat v dveh letih) izvede nenapovedano presojo, da preveri, kako imetnik certifikata vzdržuje in obnavlja sistem vodenja kakovosti. Celoten postopek certificiranja po uredbi MDR je čisto nov postopek, vendar se lahko deloma prilagodi, če je organizacija že imetnik veljavnega certifikata ES po direktivi MDD – v primeru presoje po Prilogi IX ali Prilogi XI, del A, se izvede kombinirano presojo po MDD in MDR.
Več o postopku.