Direktiva za medicinski proizvodi (93/42/EEC)
Direktiva 93/42/EEC
Direktiva 93/42/EEC za medicinske proizvode uređuje područje medicinskih proizvoda za humanu upotrebu i obuhvaća neaktivne medicinske proizvode, neaktivne implantate, proizvodi za zbrinjavanje povreda, neaktivna stomatološka pomagala, medicinske proizvode vezane na izvor napajanja/aktivna medicinski proizvodi, medicinski proizvodi za slikovnu dijagnostiku i terapiju, medicinski proizvodi za nadzor/monitoring, medicinski proizvodi za radio/termoterapiju, sterilni medicinski proizvodi. Opseg.
Zašto dobiti certifikat
Zakonodavstvo EU (93/42/EEC) u vezi s medicinskim proizvodima utvrđuje da proizvođači medicinskih proizvoda moraju označavati svoje proizvode znakom CE, prije nego što ih ponude na tržištu EU. Time izjavljuju da je medicinsko proizvod u skladu s propisima EU koji uređuju područje medicinskih proizvoda i osiguravaju da je njihov medicinski proizvod siguran i tehnički usklađen. Kada je medicinsko proizvod klase Is, Im, IIa, IIb i III, potrebno je u postupak utvrđivanja usklađenosti medicinskog sredstva uključiti i notifikacijsko tijelo (SIQ). Usklađenost sa zahtjevima propisa EU koji uređuju medicinski proizvodi se iskazuje dodjelom EC certifikata (EC Certificate).
Sustav upravljanja kvalitetom prema ISO 13485 i direktiva 93/42/EEC
ISO 13485 je harmonizirana norma kojom proizvođači medicinskih proizvoda dokazuju usklađenost sustava kvalitete u skladu sa zahtjevima direktive 93/42/EEC i, pored zahtjeva norme, proizvođači moraju uključiti i posebne zahtjeve koje utvrđuje direktiva. Pri postupku ocjenjivanja usklađenosti s Aneksima II, V i VI, preporučujemo da proizvođač ima uspostavljen i sustav kvalitete prema normi ISO 13485.
Certifikacija medicinskih proizvoda prema direktivi 93/42/EEC
Medicinski proizvodi mogu se ponuditi na tržištu EU i nositi oznaku CE ako ispunjavaju bitne zahtjeve direktive za medicinski proizvodi 93/42/EEC, a na tržištu Republike Hrvatske moraju ispunjavati zahtjeve zakona o lijekovima i medicinskim sredstvima i podzakonskih akata koje iz njega proizlaze.
Medicinski proizvodi se u odnosu na stupanj rizika dijele na:
- klasa I – medicinski proizvodi s niskim stupnjem rizika za korisnika,
- klasa IIa – medicinski proizvodi s većim stupnjem rizika za korisnika,
- klasa IIb – medicinski proizvodi s visokim stupnjem rizika za korisnika i
- klasa III – medicinski proizvodi s najvišim stupnjem rizika za korisnika.
Postupak certifikacije se sastoji od provjere dokumentacije/tehničke mape medicinskog proizvoda i od certifikacijske provjere. Preporučuje se da proizvođač medicinskih proizvoda ima uspostavljen sustav kvalitete prema normi ISO 13485. Provjera prema medicinskoj direktivi može se kombinirati s provjerom prema normi ISO 13485. Nakon dodjele EC certifikata i certifikata prema normi ISO 13485, jedanput godišnje provjeravamo djelovanje sustava redovnim provjerama u određenim dijelovima sustava, a jedanput u tri godine provodimo recertifikacijsku provjeru. EC certifikat je važeći sve dok godišnjim provjerama uspijevate dokazati da ispunjavate zahtjeve direktive 93/42/EEC.
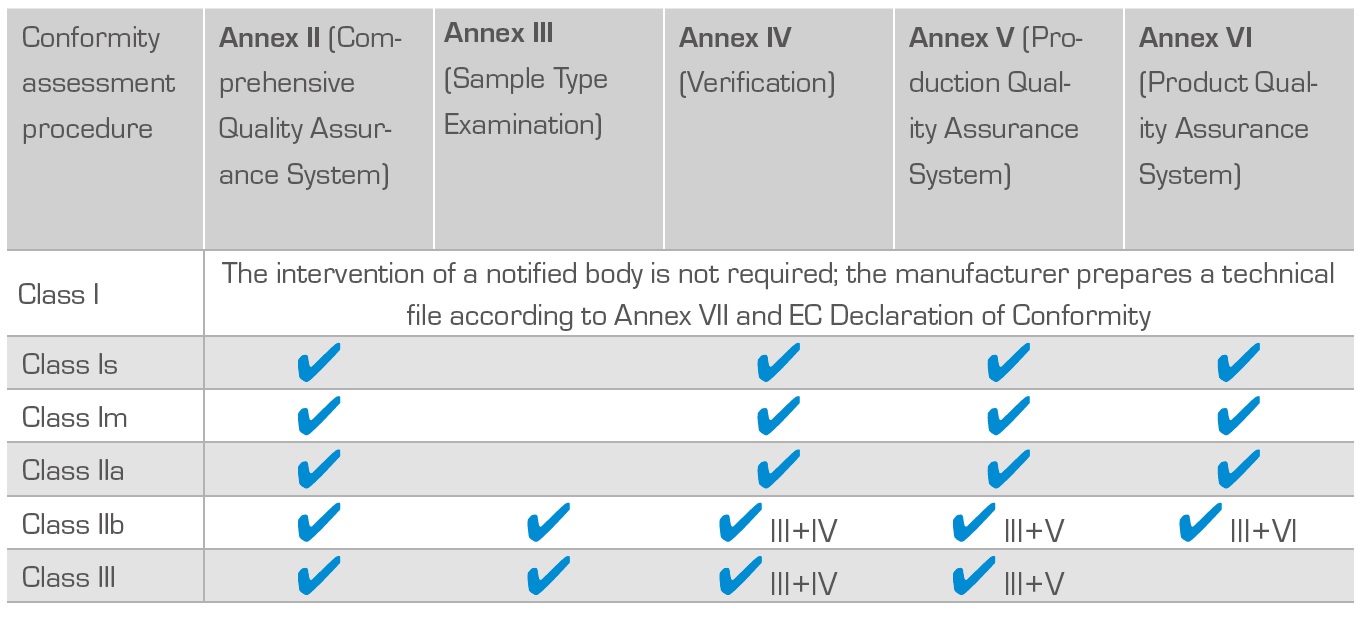
Postupci ocjenjivanja usklađenosti
Postupak ocjenjivanja usklađenosti medicinskog sredstva zavisi od klasifikacije medicinskog sredstva/klase i s time povezanim stupnjem rizika za korisnika, kao i od odluke proizvođača. Većina proizvođača se odlučuje za postupak ocjenjivanja usklađenosti prema Aneksu V ili Aneksu II.
Postupci ocjenjivanja usklađenosti medicinskih proizvoda:
Glavni koraci u postupku dobivanja CE znaka
- Klasifikacija medicinskog sredstva
- Uspostavljanje kontakta s notifikacijskim tijelom za medicinski proizvodi klase Is, Im, IIa, IIb i III
- Izbor odgovarajućeg postupka ocjenjivanja usklađenosti
- Odluka o uspostavljanju sustavu kvalitete po ISO 13485 za proizvođače medicinskih proizvoda klase Is, Im, IIa, IIb i III (s izuzetkom postupka ocjenjivanja usklađenosti prema aneksima III i IV)
- Ispunjavanje bitnih zahtjeva direktive
- Priprema tehničke mape s EC izjavom o usklađenosti
- Ocjenjivanje usklađenosti
- Stavljanje CE znaka na medicinsko proizvod
Tehnička dokumentacija/Tehnička mapa
Proizvođač mora u postupku ocjenjivanja usklađenosti pripremi ti tehničku mapu za medicinsko proizvod/grupu medicinskih proizvoda, po kojemu se provjerava ispunjavanje bitnih zahtjeva medicinske direktive. Obavezan sadržaj tehničke mape je definiran u smjernici NBOG’s BPG 2009-1. Tehnička mapa ocjenjuje notifikacijsko tijelo, osim u slučaju medicinskih proizvoda klase I, kada to nije obavezno.
Ispitivanje električne i magnetne sigurnosti za aktivna medicinski proizvodi
Proizvođači aktivnih medicinskih proizvoda u postupku ocjenjivanja usklađenosti moraju dokazati i ispunjavanje bitnih zahtjeva iz područja električne i magnetne sigurnosti. SIQ nudi priznate i akreditirane ispitne laboratorije za ispitivanje nekih vrsta električnih medicinskih proizvoda i opreme i ocjenu elektromagnetne kompatibilnosti.