Back to medical devices
Certification procedure according to Regulation (EU) 2017/745
Main steps in the process of obtaining the CE mark
- Classification of a medical device
- Establishing a contact with a notified body for medical devices of Class Is, Im, Ir, IIa, IIb and III
- Selection of the appropriate conformity assessment procedure (depending on the classification of the medical device / rule and the associated level of risk to users and the manufacturer’s decision)
- Decision to establish a quality system according to ISO 13485 for manufacturers of medical devices in Classes Is, Im, Ir, IIa, IIb and III (with the exception of the conformity assessment procedure under Annexes X and XI Part B)
- Compliance with the General safety and performance requirements of the MDR Regulation
- Preparation of the technical file (Annex II and Annex III) with the EU declaration of conformity (Annex IV MDR)
- Conformity assessment
- Attaching the CE mark to medical devices
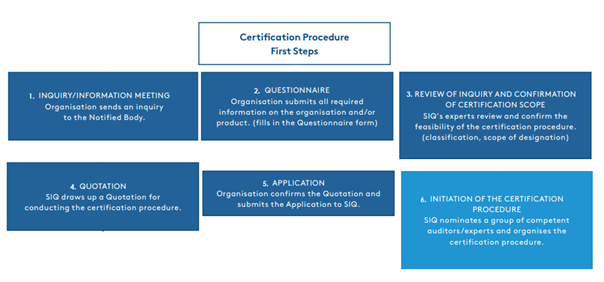
The path from establishing contact with the notified body to the start of the certification process.
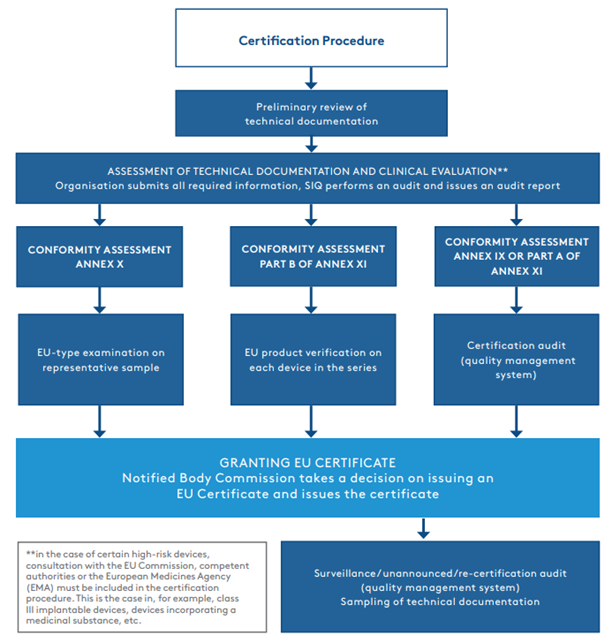
The first part of the procedure is the assessment of the technical documentation and the clinical evaluation of the device. Once the assessment of the technical documentation and the clinical evaluation has been completed and the documentation is in compliance with the MDR, the certification procedure continues according to the chosen conformity assessment procedure.
Most often, manufacturers decide on a conformity assessment procedure set out in Annex IX or Part A of Annex XI, which involves an audit of the established quality management system. In such cases, the certification procedure continues with a certification audit, which may be combined with an audit according to the ISO 13485:2016 standard. EU certificate is valid for 5 years. Once a year, we check the compliance with the quality management system with surveillance audits.